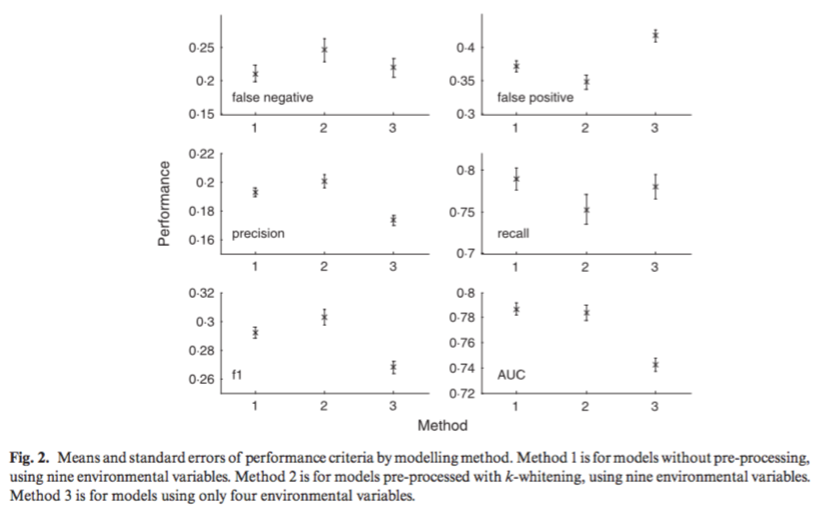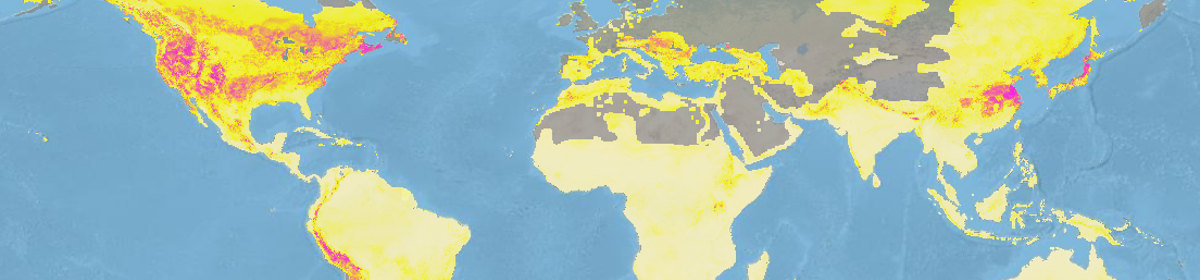
Sohanna, A. & Thomas, K., 2015. Looking Forward by Looking Back: Using Historical Calibration to Improve Forecasts of Human Disease Vector Distributions. Vector-Borne and Zoonotic Diseases, 15(3), pp.173–183. link
With rising concerns about how environmental change impacts disease vector distributions, many studies aim to predict future vector distributions under varying climate change scenarios using information available at present time. Many types of species distribution models enable us to produce highly accurate present-day data on vectors of disease. However, when trying to forecast or ‘hindcast’ species distributions many models are never validated with independent data on past or separately observed distributions. This review paper focuses on (1) methods of validation for present day spatial models, (2) how these models should be projected into the future, and (3) introduce the method of historical calibration for validation. The authors explain three methods of validation for present day spatial models and their limitations: the commonly used split-data approach (training & test data), independent dataset validation (geographically or temporally distinct data sets for validation), and validation via occurrence of disease in reservoir species. Next, the authors reviewed the use of GCMs to model future climates and their limitations including ignoring biological processes and non-linearities as well as using constant change environmental increments without setting theoretical limitations. Lastly, they suggest that historical calibration, a validation method rooted in macroecology, is more temporally transferrable in the context of projecting vector distributions and when coupled with reliable ensemble models could reduce current shortcomings in forecasting species distributions.